آموزش ساخت فایل ورودی برای نرمافزار محاسباتی گمس (GAMESS)
- توضیحات
- منتشر شده در یکشنبه, 25 آبان 1393 19:36
- نوشته شده توسط حسین حجی آبادی
- بازدید: 13154
![]()
گمس (GAMESS) نرمافزاری جامع برای انجام محاسبات کوانتومی است. این نرمافزار ازنظر عملکرد شباهت بسیار زیادی به نرمافزار گوسین (Gaussian) دارد و اغلب بهعنوان جایگزین رایگان و غیرتجاری گوسین مطرح میشود. گمس طیف گستردهای از نظریهها و روشهای محاسبات ساختار الکترونی را شامل میشود و بر روی بسیاری از پلتفرمها قابلنصب است. از دیدگاه تاریخی گمس یکی از اولین نرمافزارهای محاسبات کوانتومی است که از انحصاری شدن روشهای محاسباتی توسط شرکت گوسین جلوگیری کرده است. در این متن ابتدا تاریخچه نرمافزارهای گوسین و گمس بهصورت مختصر آورده شده و سپس ساخت فایل ورودی گمس آموزش دادهشده است.
در اواخر دهه هفتاد و اوایل دهه هشتاد میلادی، جان پوپل و همکارانش در دانشگاه کارنگی-ملون ایجاد نرمافزاری جامع برای انجام محاسبات ساختار الکترونی را آغاز کردند. اولین نسخه این نرمافزار که بر اساس توابع پایه گوسی ساختهشده بود، گوسین 70 نامیده شد. گوسین 70 و نسخههای بعدی آن تا گوسین 80 بهصورت رایگان در اختیار عموم قرار میگرفت. بعدازاین نسخه بهمنظور تأمین بخشی از هزینههای توسعه این نرمافزار مبلغ اندکی از استفادهکنندگان نرمافزار دریافت میشد. این سیاست باعث شد تا توسعه نرمافزار گوسین بهصورت تجاری درآید و شرکت گوسین (Gaussian, Inc) بدین منظور تأسیس شد. شرکت گوسین بهتدریج از اهداف علمی اولیه پدیدآورندگان این نرمافزار دور و به سمت درآمدزایی بیشتر حرکت کرد. پروفسور پوپل دانشمند برنده جایزه نوبل شیمی، به همکاری خود را با شرکت گوسین در سال 1991 پایان داد.
از آن تاریخ تاکنون، شرکت گوسین هزینه بالایی را از استفادهکنندگان این نرمافزار دریافت میکند. همچنین مجوز گوسین قوانین بسیار سختگیرانهای را به کاربرانش اعمال میکند. بهعنوان نمونه کاربران گوسین حق مقایسه کارایی و سرعت انجام محاسبات گوسین با نرمافزارهای مشابه را ندارند! بر اساس این قوانین بسیاری از دانشگاهها و اشخاص مهم توسط شرکت گوسین تحریم شدهاند. تعدادی از این دانشگاهها و اشخاص در این لینک، نامبرده شدهاند. نکته جالب و درعینحال تأسفبرانگیز دراینباره این است که خود پروفسور پوپل جزء کسانی بود که در زمان حیاتش توسط شرکت گوسین تحریم شده بود و اجازه استفاده از آن را نداشت. تجاری شدن و سختگیریهای شرکت گوسین باعث شد تا بسیاری از دانشگاههای معتبر و بزرگان شیمی محاسباتی به فکر جایگزینی برای این نرمافزار محاسبات ساختار الکترونی بیفتند. خوشبختانه چنین نرمافزار جایگزینی از قبل وجود داشت. کد نویسی پروژه گمس در سال 1977 و قبل از تجاری شدن گوسین شروعشده بود. در سال 1981 این پروژه به دو بخش GAMESS(US) و GAMESS(UK) تفکیک شد. بخش آمریکایی گمس از آن تاریخ تاکنون در دانشگاه ایالت آیوا نگهداری و توسعه داده میشود. رایگان بودن و مجوز غیرتجاری گمس US باعث شد تا بسیاری از محققان برای رهایی از انحصارطلبی شرکت گوسین به سمت این نرمافزار گرایش پیدا کنند.
در کشور ما متأسفانه استفاده از کپی غیرقانونی نرمافزارها متداول است. هرچند کاربران گوسین در ایران هزینهای بابت مجوز این نرمافزار پرداخت نمیکنند و برای استفاده غیرقانونی از آن مورد پیگرد قانونی قرار نمیگیرند ولی بااینحال استفاده از این نسخههای غیرقانونی میتواند مشکلاتی برای آنها به همراه داشته باشد. بهعنوانمثال بسیاری از مجلات علمی، کارهایی را که با نسخه غیرقانونی نرمافزارهای علمی انجامشده باشند را نمیپذیرند. مشکل دیگر استفاده از نرمافزارهای بدون مجوز عدم پشتیبانی فنی از آنهاست. پشتیبانی فنی خوب نیمی از ارزش یک نرمافزار علمی است. درصورتیکه کاربران نرمافزارهای بدون مجوز با مشکلی مواجه شوند، از سازنده نرمافزار هیچ کمکی دریافت نمیکنند. کاربران گوسین در ایران میتوانند با جایگزینی نرمافزار گمس از مشکلات استفاده از یک نرمافزار بدون مجوز رهایی یابند. مزیت دیگر استفاده از گمس بروز بودن آن است. نرمافزار گمس در هرسال بین یک تا دو بار بهروز میشود، بنابراین کاربران آن میتوانند از بروزترین روشهای محاسباتی استفاده کنند. این در حالی است که تمامی نسخههای گوسین موجود در ایران تاریخ گذشته هستند. در ادامه آموزش ساخت فایل ورودی گمس ارائهشده است. نگارنده این متن امیدوار است تا استفاده از این نرمافزار کارآمد، بروز، قانونی و رایگان در ایران رواج بیشتری پیدا کند.
آشنایی با قالب فایل ورودی گمس
مانند تمامی نرمافزارهای محاسباتی، فایل ورودی گمس یک فایل ساده متنی است که میتوان آن را با استفاده از یک ویرایشگر ساده متنی مانند vi در محیط یونیکس/لینوکس یا notepad در محیط ویندوز تهیه کرد. برای تهیه این فایل باید قالب خاص آن را رعایت کرد در غیر این صورت اجرای محاسبات با خطا مواجه خواهد شد. ورودی گمس از گروههای مجزایی تشکیلشده است که هر گروه اطلاعات خاصی را از طریق کلمات کلیدی مخصوص به خود به نرمافزار منتقل میکند. گروههای گمس با عبارت $GroupName شروع میشود و در پایان با عبارت $END خاتمه مییابد. مهمترین گروههای فایل ورودی گمس عبارتاند از:
$SYSTEM، مقدار حافظه مورداستفاده گمس و محدودیت زمانی اجرای محاسبه را تعیین میکند.
$CONTRL، پارامترهای اساسی درباره نوع چگونگی انجام محاسبه را تعیین میکند.
$BASIS، توابع پایه استاندارد موردنیاز محاسبه را تعیین میکند.
$DATA، مختصات مولکولی و توابع پایه غیراستاندارد را تعیین میکند.
گروههای مهم دیگری که بهطورمعمول در یک فایل ورودی وجود دارند عبارتاند از:
$GUESS، این گروه مشخص میکند که اوربیتال مولکولی اولیه (برای شروع محاسبات SCF) چگونه حدس زده شود.
$SCF، پارامترهای مربوط به محاسبه تابع موج را تعیین میکند.
$FORCE، این گروه چگونگی محاسبه ماتریس هسی (ماتریس مشتق دوم انرژی که بهعنوان ماتریس ثابت نیرو نیز شناخته میشود) را تعیین میکند. پارامترهای این گروه اغلب برای محاسبه بهینهسازی ساختار موردنیازند.
$HESS، این گروه مشخص میکند که ماتریس هسی اولیه چگونه حدس زده شود.
$IRC، این گروه پارامترهای مربوط به محاسبه مسیر واکنش را تعیین میکند.
بهجز این گروهها، فایل ورودی نرمافزار گمس میتواند حاوی بسیاری از گروههای دیگر باشد که هرکدام پارامترهای مربوط به یک محاسبه خاص را تعیین خواهند کرد. برای مشاهده لیست کامل گروههای گمس میتوانید از دستور gmshelp در محیط یونیکس/لینوکس استفاده و یا فایل input.pdf را از وبسایت گمس دانلود کنید. همانطور که در این منابع مشاهده خواهید کرد، پارامترهای بسیار زیادی توسط کلمات کلیدی درون گروهها تعیین میشوند. بااینحال نگران تعداد زیاد پارامترها نباشید. بسیاری از کلمات کلیدی دارای مقادیر پیشفرض هستند بنابراین لازم نیست تا کاربر تمام پارامترها را تعیین کند. $basis و $data تنها گروههایی فاقد مقدار پیشفرضاند و حتماً باید توسط کاربر تعیین شوند.
برای قرار دادن گروهها در فایل ورودی گمس چند قاعده اساسی وجود دارد که عبارتاند از:
- علامت $ ابتدای هر گروه تنها میتواند در ستون دوم قرار بگیرد. علامت $ انتهایی میتواند در هر ستونی بهجز ستون اول قرار داده شود.
- اگر کاراکتری در ستون اول یک خط قرار گیرد، گروهی که درون آن خط وجود دارد، توسط نرمافزار خوانده نمیشود. در مثال دوم خطی که با علامت! در ستون اول شروعشده، صرفاً برای یادداشتگذاری نوشتهشده و توسط برنامه خوانده نمیشود.
- یک خط در ورودی گمس نمیتواند بیشتر از 80 ستون داشته باشد. عباراتی که فراتر از این حد قرار گیرند خوانده نخواهند شد. بااینحال میتوان یک گروه را در بیش از یک خط قرارداد.
- ترتیب قرار گرفتن گروهها در فایل اهمیتی ندارد.
- استفاده از حروف بزرگ و کوچک اهمیتی ندارد. بهعنوانمثال عبارتهای زیر کاملاً یکسان هستند:
$CONTRL RUNTYP=OPTIMIZE DFTTYP=B3LYP $END
$Contrl RunTyp=OptimiZe DFTTYP=B3lyp $End
$contrl runtyp=optimize dfttyp=b3lyp $end
- چند نمونه از یک گروه میتواند در یک فایل ورودی وجود داشته باشد که در این صورت تنها آخرین نمونه گروه در نظر گرفته خواهد شد. بهعنوانمثال اگر دو گروه $contrl به ترتیب زیر در یک فایل وجود داشته باشد. یک محاسبه بهینهسازی ساختار (optimization)انجام خواهد شد.
$contrl runtyp=energy dfttyp=b3lyp $end
$contrl runtyp=optimize dfttyp=b3lyp $end
- ترتیب کلمات کلیدی در داخل یک گروه اهمیتی ندارد. بنابراین دو خط زیر باهم برابر هستند:
$contrl runtyp=optimize dfttyp=b3lyp $end
$contrl dfttyp=b3lyp runtyp=optimize $end
بهعنوان نمونه به ورودی زیر توجه کنید که برای انجام محاسبه انرژی روی مولکول هیدروژن نوشتهشده است. این فایل ورودی تنها از سه گروه $contrl، $basis و $data تشکیلشده است.
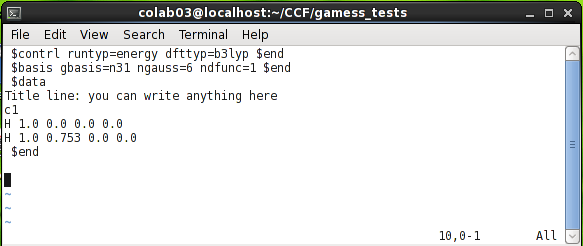
در این فایل عبارت runtyp=energy در گروه $control مشخص میکند که یک محاسبه انرژی باید صورت گیرد. محاسبات دیگری که با استفاده از این کلمه کلیدی میتوان درخواست کرد، عبارتاند از: Gradient، برای محاسبه گرادیان انرژی، Optimize، برای محاسبه ساختار بهینه، Hessian، برای محاسبه مشتق دوم انرژی، فرکانسهای ارتعاشی و خواص ترمودینامیک، Sadpoint، برای جستوجوی نقطه زینی، IRC، برای محاسبه مسیر ذاتی واکنش. پسازآن، عبارت dfttyp=b3lyp مشخص میکند که از روش DFT و تابع b3lyp استفاده شود. گروه $control محدود به این دو کلمه کلیدی نمیشود و میتواند شامل عبارات دیگری نیز باشد که مهمترین آنها عبارتاند از:
Icharge: برای تعیین بار سیستم. مقدار پیشفرض برابر با صفر است.
mult: برای تعیین چندگانگی سیستم. مقدار پیشفرض برابر با یک است.
exetyp: این کلمه کلید مشخص میکند که آیا یک محاسبه واقعی انجام شود و یا فقط فایل ورودی چک شود. مقادیر مجاز عبارتاند از: check و run
units: واحد فایل ورودی را مشخص میکند که میتواند برابر angs (مقدار پیشفرض) و یاbohr باشد.
scftyp، برای مشخص کردن نوع تابع موج (rhf، rohf، uhf، mcscf و یا GVB)
mplev: برای مشخص کردن مرتبه نظریه اختلال. مقدار پیشفرض برابر با صفر است.
cctyp: برای استفاده از روش coupled cluster
گروه $basis نوع توابع پایه مورداستفاده را مشخص میکند. در این گروه عبارت gbasis=n31 مشخص میکند که یک مجموعه تابع پایه گوسی از نوع n31 مورداستفاده قرار میگیرد. عبارت ngauss=6 تعداد توابع گوسی مورداستفاده برای هسته را برابر با 6 قرار میدهد. عبارت ndfunc=1 مشخص میکند که از یک اوربیتال d اضافی برای بهتر لحاظ شدن قطبش پذیری مولکول استفاده شود. بهصورت کلی تابع پایهای که گروه $basis درخواست میکند 6-31g(d) است.
گروه $data در فایل نمونه بالا، مختصات مولکولی مشخص کرده است. در نمونه از مختصات کارتزین با قالب زیر برای مشخص کردن مکان اتمها استفادهشده است.
>Symbol Z x_coord y_coord z_coord
که symbol نوع اتم، Z با هستهاتم و مابقی خط مختصات اتم در فضای دکارتی را مشخص میکند. گروه $data بهجز مختصات مولکولی حاوی اطلاعات دیگری نیز است. عبارت c1 در بالای مختصات مولکولی نوع گروه نقطهای ساختار را مشخص میکند و خط title میتواند حاوی عنوان محاسبه باشد.
نمونهای پیچیدهتر از فایل ورودی گمس در شکل زیر نشان دادهشده است. این ورودی یک محاسبه بهینهسازی ساختار را برای یون هیدرونیم از گمس درخواست میکند. این محاسبه با استفاده از روش mp2 و تابع پایه 6-311g(d,p) انجام خواهد شد. توجه کنید که به خاطر قرار گرفتن یک کاراکتر در ستون اول، خط اول این فایل توسط نرمافزار خوانده نخواهد شد و از آن تنها برای یادداشت نویسی استفادهشده است. همچنین گروه $contlr به علت طولانی بودن در دو خط تقسیمشده است.
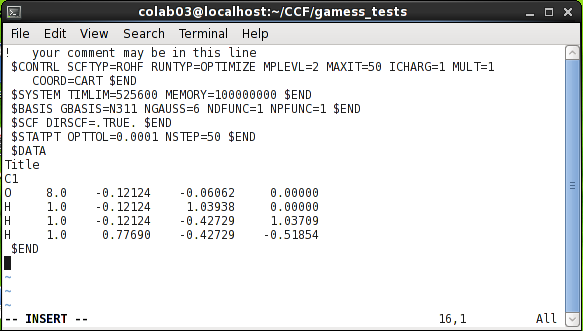
در داخل گروه $system دو کلمه کلیدی وجود دارد. عبارت TIMLIM=525600 محدودیت زمانی اجرای محاسبه را برحسب ثانیه تعیین میکند. درصورتیکه محاسبه بعدازاین زمان کامل نشود، گمس اجرای آن را بهصورت ناتمام خاتمه خواهد داد؛ و عبارت memory=100000000 حافظه در دسترس نرمافزار را برحسب word (هر word برابر با 8 byte است)تعیین میکند.
در داخل گروه $scf، عبارت DIRSCF=.TRUE. باعث میشود تا گمس بهجای خواندن انتگرالها از روی دیسک، در هر مرحله آنها را دوباره محاسبه کنید. این کلمه کلیدی در بسیاری از رایانهها موجب افزایش قابلتوجه سرعت محاسبات خواهد شد.
گروه $statpt شامل کلمات کلیدی است که چگونگی جستوجوی ساختار بهینه و نقطه زینی را تعیین میکنند. در مثال بالا کلمات کلیدی opttol و nstep به ترتیب میزان آستانه همگرا شدن گرادیان انرژی و حداکثر تعداد سیکلهای بهینهسازی را مشخص میکنند.
ساخت فایل ورودی با استفاده از نرمافزار macmolplt
Macmolplt نرمافزار گرافیک مولکولی است که برای مشاهده و نتایج و ساخت فایل ورودی گمس طراحیشده است. هرچند این نرمافزار امکانات زیادی برای طراحی ساختارهای مولکولی ندارد ولی فایل ورودی گمس را بهخوبی میشناسد و با استفاده از آن میتوان یک فایل ورودی با تنظیمات مناسب تهیه کرد. برای ایجاد یک فایل ورودی با استفاده از این نرمافزار باید روی منوی subwindow←input Builder کلیک کنید.
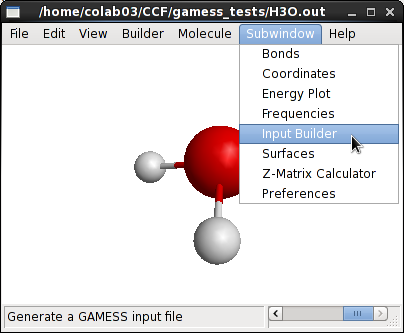
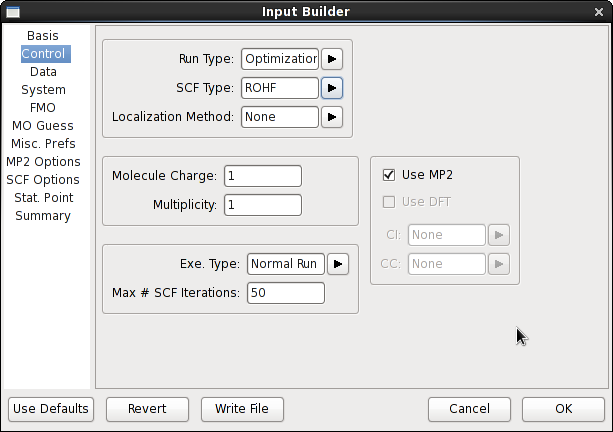
در سمت چپ پنجره input Builder لیست انواع گروههای مختلف وجود دارد. پس از انتخاب نوع گروه میتوانید پارامترهای مختلف گروه مربوطه را در سمت راست پنجره تغییر دهید. در پایان دکمه Write File کلیک کنید تا فایل ورودی ایجاد شود.
ساخت مختصات مولکولی برای ورودی گمس با استفاده از gaussview
بسیاری از کاربران نرمافزارهای محاسباتی با نرمافزار gaussview آشنا هستند. این نرمافزار امکانات بسیار خوبی برای طراحی ساختارهای شیمیایی دارد. برای استفاده از مختصات مولکولی ساختهشده با استفاده از gaussview در ورودی گمس، ابتدا مختصات مولکولی را با فرمت mol و یا pdb ذخیره کنید. پسازآن میتوانید این فایل را با استفاده از نرمافزار open babel به ورودی گمس تبدیل کنید. برای تبدیل فایل در محیط یونیکس از میتوانید از رابط گرافیکی open babel استفاده کنید و یا دستور زیر را در محیط متنی اجرا کنید:
obabel input.mol –O input.inp
دیدگاهها
سلام
خیر، تاجایی که من اطلاع دارم. منبع فارسی نداره!
سلام
برای دریافت گمس ابتدا در این قسمت آدرس ایمیل و نسخه مورد نظر خود را وارد کنید:
http://www.msg.ameslab.gov/GAMESS/download/register/
سپس با استفاده از نام کاربری، رمز و لینکی که برای شما ارسال می شود گمس را دانلود کنید.
من موقع دانلود در هنگام وارد کردن رمز با مشکل مواجه می شوم!?
error:expecting an even number of MPI processess مواجه میشم
چه راه حلی رو پیشنهاد می دهید ؟
با تشکر
سلام
اول اینکه شما exetyp=check قرار دادید بنابراین برنامه تنها فایل ورودی شما رو چک میکنه و محاسبه انجام نمیشه
دوم اینکه دوبار از کیبورد runtyp استفاده کردید. ابتدا ساختار رو بهینه کنید بعد یک محاسبه دیگه برای محاسبه فرکانس انجام بدید. در آخر اگه باز هم به این خطا برخوردید تعداد سیکل های scf رو با قرار دادن کیبورد زیر در گروه contrl افزایش بدید: MAXIT=N
را نوشتم تا مانند نرم افزار گوسین opt و freqانجام دهدبه شرح ذیل
INPUT CARD> $CONTRL SCFTYP=ROHF EXETYP=CHECK RUNTYP=OPTIMIZE RUNTYP=HESSIAN DFTTYP=B3LYP
INPUT CARD> ICHARG=-4 MULT=2 COORD=ZMT
و...
اما با اینکه خروجی آن نرمال به پایان رسید ولی چنین خطایی رانیز داد
THE VIBRATIONAL ANALYSIS IS NOT VALID
THE SCF COMPUTATION DID NOT CONVERGE AT ONE OR MORE OF THE DISPLACED GEOMETRIES. YOU SHOULD CONVERGE RUN'S -RESTART- FILE, THEN RESTART THIS JOB
سلام.
روش CAM-B3LYP مثل خود روش B3LYP یک روش DFT هیبریدی است با این تفاوت که برای مطالعه charge transfer excitations بهینه سازی شده. برای اطلاعات بیشتر مقاله زیر را مطالعه کنید:
T. Yanai, D. Tew, and N. Handy, “A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP),” Chem. Phys. Lett., 393 (2004) 51-57.